

Flow Cytometry Basics
Getting Started
Flow cytometry can be useful for making a number of measurements about a cell. In
its most simplistic form forward and side scatter can indicate basic characteristics
such as size and granularity, but what about cell type, activation, or expression
level? Obviously, flow cytometry can be very limited unless we take full advantage
of excitation and emission of fluorescent molecules.
In order to do this, cells need to be prepared for either analysis or sorting using
antibodies or fluorescent probes. This generally encompasses a few steps depending
on the source tissue or cell type, markers of interest, marker location in or on the
cell, and cell process. Always remember that it is very important that you know exactly what you want to measure and in which population of cells you want to measure it.
So, prior to using the Flow Cytometry Core Facility, whether you intend to sort or
analyze, we ask that you be aware of a few rules regarding sample submissions. We've
also provided here a set of basics regarding reagents to use, sample preparation,
blocking, permeabilization, and staining as well as a few basic protocols to get you
started in the right direction.
Rules and Regulations Regarding Samples
Generally
All samples should be prepared by the individual investigator or researcher. Staining
will not be performed by the Core Facility. However, the core facility will provide
assistance and guidance with experimental design, controls, seeding of samples, preparation
for sorting and analysis, and answers to general questions regarding aspects of flow
cytometry.
Cancellation of appointments with at least a 24-hour notice will not be charged. Failure
to notify of cancellation within 24 hours will result in billing for the time requested.
Special circumstances and considerations will be noted.
Samples of Pathogenic Potential
It is the policy of the Core Facility that all information about the kind of samples being run at the facility, with or without assistance, be disclosed. The facility must be aware of any samples containing pathogenic potential such as transforming viruses, unfixed bacteria known to cause disease, etc. at the time of scheduling. The aerosol management system used for containment may be used for such samples during the sort process to reduce contamination of the equipment or protect personnel using the equipment. Users who do not disclose pertinent information relating to samples will not be allowed to schedule appointments. Repeated non-disclosure will result in loss of privileges and use of the Core Facility for a period of time. Please plan accordingly.
Human Samples
Human samples must be pre-screened for the following: HIV, Hepatitis B and Hepatitis
C. The Core Facility must be notified at the time of scheduling if these samples are
used (see above). Non-screened human samples will not be sorted; no exceptions will
be made. Human samples that are fixed can be analyzed.
Human samples must have the source of tissue identified. The individual investigators
who use pre-screened human tissue must provide an identification number or another
means of identifying the source of the tissue in accordance with HIPAA regulations.
Sample ID and other pertinent information must be known by the individual investigator
and Core Facility personnel as a way to identify the source of the tissue in case
of an emergency and will be kept for future records.
Self-Operation
Investigators, students, technicians, and other interested individuals may be allowed to run the FACSCalibur themselves. These self-operation users must be trained on the FACSCalibur by Core personnel or must have had training on the FACSCalibur platform prior to use of the instrument. Training will be provided consisting of 1-2 sessions, or initial training, that provide basic information relating to general flow cytometry, machine operation, and maintenance. No samples should be brought to the first session. A second session will conclude training with researcher-provided sample analysis and user operation of the machine with minimal supervision/assistance from Core personnel. Only training for the FACSCalibur will be provided at this time. The FACSAria will be solely operated by Core Facility personnel and certified users.
Do You Need a Whole New Set of Reagents?
Before you set off in the bold new direction of flow cytometry, it's best to make sure that you have all the necessary reagents on hand. This includes dissociative agents, permeabilization/fixation solutions, antibodies, viability dyes, blocking reagents, and buffers for blocking, staining, analyzing, or sorting. It may seem like there's a lot, but most of the necessities are probably already in your lab.
Dissociatives
Dispase
- Dispase® II
- NaHCO3
- HBSS without Calcium or Magnesium
- HEPES Buffer, 1M
Dissolve 0.3g of dispase into 75mL of HBSS. Add 2.5mL of HEPES (25mM final concentration) to the solution and mix well. Adjust the pH to ~7-7.2, filter sterilize the solution, and aliquot into appropriate volumes. This is stable at 2-8°C for ~3 days and longer if kept at -22°C.
For use in preparing single-cell suspensions from some tissues and cell cultures. This may not be suitable for your particular cell type; so, before preparing check current literature for particulars.
Trypsin-EDTA, 0.25%
Permeabilization
- Dispase® II (dry)
- NaHCO3
- HBSS without Calcium or Magnesium
- HEPES Buffer, 1M
Dissolve 0.5g of dispase into 75mL of HBSS. Add 2.5mL of HEPES (25mM final concentration) to the solution and mix well. Adjust the pH to ~7-7.2 and filter sterilize the solution and aliquot into appropriate volumes. This is stable at 2-8°C for ~3 days.
Sample Running/Sorting Buffer
- HBSS without Calcium or Magnesium
- Foetal Bovine Serum (FBS) or Bovine Serum Albumin (BSA)
- HEPES, 1M
- EDTA, 5M
- DNAse I
- MgCl2, 1M
In 500mL of HBSS mix 5mL FBS (or BSA to 1%), 5mL HEPES, 0.5mL EDTA (5mM final concentration), and 2.5mL MgCl2. Add 12.5mg DNAse I (final concentration of 25ug/mL) to the solution and mix well. Adjust the pH to ~7-7.2 and filter sterilize the solution and aliquot into appropriate volumes. This is stable at 2-8°C for ~30 days.
Are Your Cells Ready For Flow?
Investigators (students, technicians, and other users) must prepare samples in their individual laboratories. Staining will not be done at the Core Facility. If needed, Core personnel can help or advise regarding sample preparation for various protocols. Contact the Core with any inquiries. All users must complete a Sample Submission Form that provides Core personnel with information needed to process an experiment. Forms can be found here. It is recommended that all investigators consult with the Core with respect to experimental design, controls, and other needs that will help meet their goals. This is to ensure that usage of the Core Facility will be extremely beneficial to all involved. The facility may accept potentially pathogenic samples for cell sorting. Should you have questions relating to human/primate or lentivirus-containing/treated samples, please consult the Core prior to scheduling appointments. Samples labeled with radioactive materials will not be accepted at any time.
The key word in flow cytometry is flow. Thus, plated cells or whole tissue cannot be assessed by flow cytometric means because they are fixed to a surface or each other. So, in order to make any use of our instrumentation, you will first need to prepare your cells in a single-cell suspension.
Is There a Need To Block?
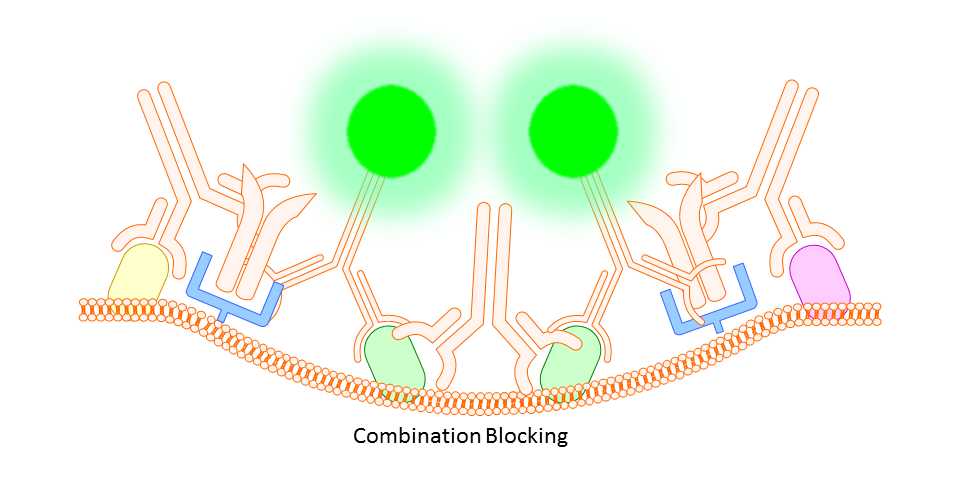
Whether you block or not is completely up to you and the protocol you choose. However, there are some situations where you will find that blocking is necessary. In some cases you might find a high degree of background fluorescence or non-specific staining in your sample. In either case, before scrapping the project all together you may find that one or both of the following work to increase specific binding and reduce background fluorescence.
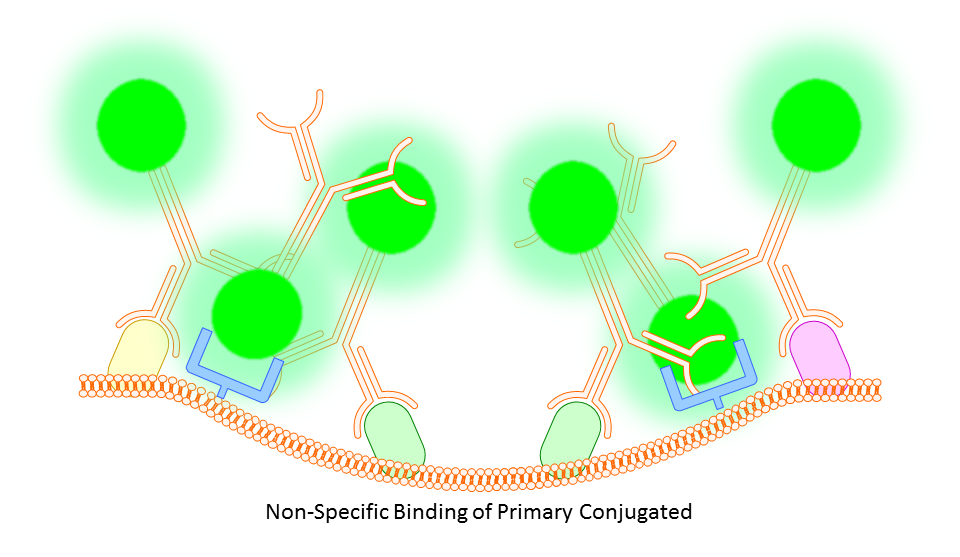
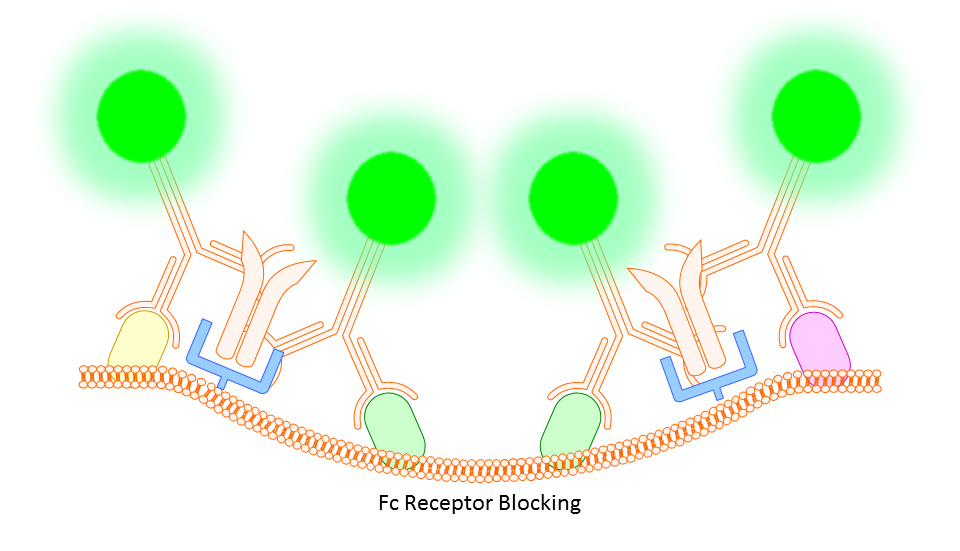
Anti-CD16/32 (Fc Receptor Block)
This targets the Fc receptor on cells in mouse or rat tissues, though generally not used or necessary for study of human cells. By binding the Fc receptors, monoclonals should be less likely to bind non-specifically resulting in better targeting of your marker.
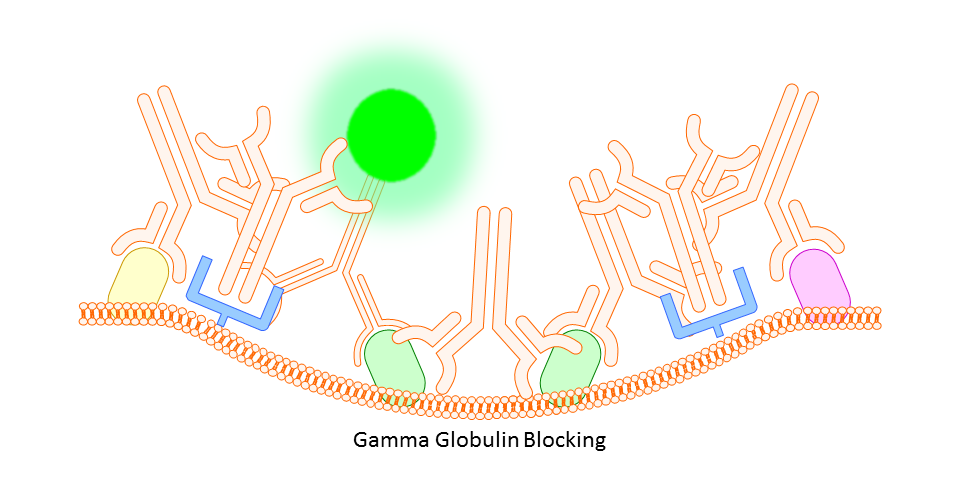
Gamma Globulin
A cheaper and often more all-encompassing method for use in both animal and human cell study is the use of unlabeled gamma globulin (IgG) as a blocking agent. Here you should use the same type of gamma globulin as host species of the primary antibody (so, if your antibody is rat IgG2a anti-mouse, use rat gamma globulin). The idea is that the gamma globulin will bind non-specifically to anything on the mouse tissue or cells that might otherwise bind your targeting antibody non-specifically, thereby reducing any background fluorescence signal.
Why Fixation/Permeabilization?
You won’t always need to fix or permeablize the cells you are interested in. sometimes the process can negatively affect surface staining (increased signal to noise or decreased signal intensity), but mostly it’s just a supercilious step except when using some intracellular or intranuclear/nucleic acid stain (many of these dyes are often impermeant, i.e. they can’t penetrate live cell or nuclear membranes) or when there is need to preserve stained cells for later analysis (this, too, can affect signal intensity and signal to noise).
Depending on the antibody and reagents being used, you will need to tailor your fixation/permeablization accordingly. Most common methods include use of 70% ethanol, absolute methanol, or 1% formalin/formaldehyde (with or without detergent). There are also some commercial products available designed specifically for certain types of intracellular staining.
Brightness and Discrimination
Surface marker staining can be performed directly by use of primary antibodies, or indirectly by using a combination of mono and poly clonal antibodies or monoclonal antibodies labeled with biotin. Primary antibody staining tends to be the most efficient and gives the clearest signal, but this is not always the case with weakly expressing cells. Often is such a case the use of indirect staining can boost weak signals; however, this does run the risk of increased background due to non-specific binding of secondary polyclonals. To avoid this, many opt to use a biotin conjugated primary antibody and then stain using a streptavidin complex. Multiple streptavidins can bind a single biotin label, and this has the effect of boosting an otherwise weak signal without the worry of non-specific binding.
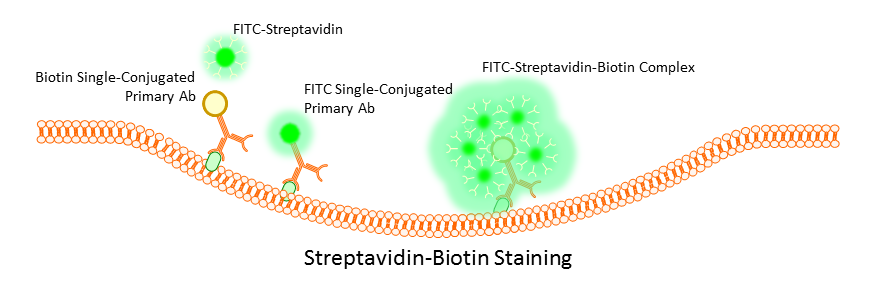
A balance will always have to be found during the staining process between concentration of the antibody used, time of incubation, and temperature. Optimization of these factors is critical to obtaining clean data. Too long, too large a concentration, or too high a temperature, and the signal may be too bright for effective data analysis, or worse, non-specific binding or cross-reactivity will occur even with a primary antibody. Use too little antibody for too short a time at too low a temperature and you will get very little to no binding and no signal. Most antibodies come with suggested starting concentrations, but more importantly, trust the experience of others in your lab or protocols that have been used by other researchers on similar instrumentation.
Most intracellular or nuclear staining is done using fluorescent molecules or fluorescently labeled peptides, etc. There’s a whole host of products depending on the application; so, feel free to ask which will work best with your current set up.
Still have questions? Check out our other basics or hit-up the contact link.